Ventilatoare Trilogy EVO: Philips lansează o alertă de siguranță, cu soluție integrată
O posibilă acumulare de particule externe în ventilatoarele Philips Trilogy Evo necesită instalarea unui filtru suplimentar, de către furnizorii de servicii medicale.
Praf, polen, reziduuri de aerosoli, toate particulele prezente în aer se pot acumula în interiorul senzorului de debit intern al ventilatoarelor Trilogy Evo și poate provoca ocluzia parțială a acestuia. Acest fenomen ar putea altera precizia parametrilor de asistență respiratorie (presiune, volum sau debit), cu posibile consecințe asupra sănătății în absența unui management adecvat.
Pentru a evita acest lucru, Philips s-a angajat să furnizeze filtre de particule specifice, singurele compatibile cu ventilatoarele sale. Utilizarea lor, anterior opțională, devine obligatorie. Aceste filtre vin în plus față de filtrul de admisie a aerului din spumă deja instalat. Acestea împiedică o mare parte a aerosolilor și a particulelor în aer să pătrundă în dispozitiv. Philips anunță că le are pe stoc și că este nevoie de maximum două săptămâni pentru a le duce la furnizorii de servicii medicale , care le vor instala rapid pe ventilatoare.


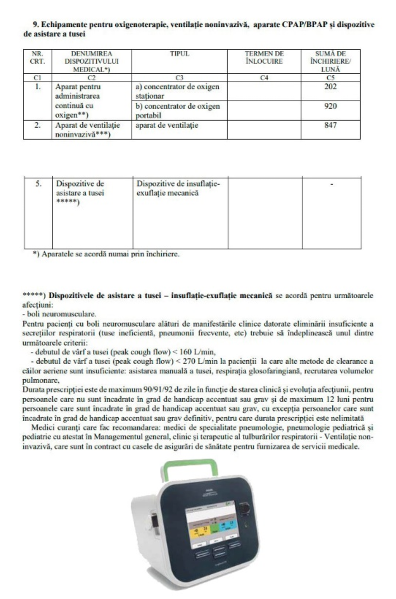
Caught Assis intra in lista dispozitivelor decontate de CNAS
Ne simțim bucuroși și recunoscători, dar și ușor mândri, pentru că, după demersuri îndelungi, am reușit să convingem factorii de decizie din CNAS de necesitate decontării prin închiriere a dispozitivului de asistare a tusei ( cough assist ) pentru pacienții cu afecțiuni neuromusculare cu afectare respiratorie.
Nu cunoaștem încă suma decontată lunar de către CNAS pentru închirierea lunară a dispozitivului dar sperăm că, costul va fi decontat în totalitate.
Ținem să mulțumim doamnei doctor Mihaela Oros și Societății Române de Pneumologie pentru sprijin..
Sursă:
https://cnas.ro/transparenta-decizionala/#0df417ec348738b02
Date prezentate la întâlnirea anuală a Academiei Americane de Neurologie desfășurată în perioada 22-27 aprilie
Tratamentele aprobate atât de agenția americană a medicamentelor ( FDA) cât și de agenția europeana ( EMA) și decontate și în România conform protocoalelor terapeutice sunt :
Terapia genică cu administrare unică Zolgensma (onasemnogene abeparvovec-xioi)
Spinraza (nusinersen), care se administrează prin injecție în coloană la fiecare patru luni după administrarea dozelor de încărcare.
Molecula cu administrare orală zilnică Everysdi (risdiplam)
Toate aceste medicamente s-au dovedit eficiente în încetinirea progresiei AMS în studiile clinice studiile desfășurate până în prezent concentrându-se în principal pe copiii mici în stadiile incipiente ale AMS.
Există mai puține date despre utilizarea acestor tratamente în populația mai largă de persoane care trăiesc cu boala – inclusiv la pacienții în vârstă și la cei la care boala este mai avansată. Date despre combinații între aceste medicamente sau trecerea de la un tratament la altul sunt, de asemenea, limitate.
JEWELFISH: 24-month Safety, Pharmacodynamic and Exploratory Efficacy Data in Non-Treatment-Naïve Patients with Spinal Muscular Atrophy (SMA) Receiving Treatment with Risdiplam
https://www.aan.com/MSA/Public/Events/AbstractDetails/54617
Spinraza in Adults with Spinal Muscular Atrophy (SAS) – 14 month results
https://www.aan.com/MSA/Public/Events/AbstractDetails/52996
The Use of Onasemnogene-abeparvovec at 33 weeks’ gestation in Spinal Muscular Atrophy with One Copy of SMN2
https://www.aan.com/MSA/Public/Events/AbstractDetails/54376
Real-world Outcomes of Nusinersen or Onasemnogene Abeparvovec (OA) Monotherapy, or Switching to OA from Nusinersen in SMA Patients Aged ≥6 Months
https://www.aan.com/MSA/Public/Events/AbstractDetails/54614
Baseline Characteristics and Interim Safety in RESPOND: A Phase 4 Study in Children with Spinal Muscular Atrophy (SMA) Treated With Nusinersen After Onasemnogene Abeparvovec
https://www.aan.com/MSA/Public/Events/AbstractDetails/54616
Long-Term Follow-Up of Onasemnogene Abeparvovec Gene Therapy in Patients with Spinal Muscular Atrophy Type 1
https://www.aan.com/MSA/Public/Events/AbstractDetails/54619
Outcomes in Patients with Spinal Muscular Atrophy and Four or More SMN2 Copies Treated with Onasemnogene Abeparvovec: Findings from RESTORE
https://www.aan.com/MSA/Public/Events/AbstractDetails/54620
Bulbar Function in Children with Two or Three SMN2 Copies Who Received Onasemnogene Abeparvovec Presymptomatically for Spinal Muscular Atrophy
https://www.aan.com/MSA/Public/Events/AbstractDetails/54621
FIREFISH Parts 1 and 2: 36-month safety and efficacy of risdiplam in Type 1 spinal muscular atrophy (SMA)
https://www.aan.com/MSA/Public/Events/AbstractDetails/54622
Treatments and Outcomes for Patients with Spinal Muscular Atrophy Type 2: Findings from RESTORE Registry
Experiența implementării la nivel național a screening-ului neonatal pentru AMS în Germania
Mai multe programe pilot, desfășurate între 2018 și 2021, în diferite regiuni din Germania, au demonstrat beneficiile screening-ului neonatal pentru atrofia musculară spinală (AMS) la peste 500.000 de nou-născuți testați. Din octombrie 2021, screening-ul la naștere pentru depistarea atrofiei musculare spinale are loc pe întreg teritoriul Germaniei.
S-a realizat un studiu privind trecerea la nivel național a screening-ului: această trecere de la programe pilot la screening la nivel național nu a afectat rapiditatea depistării sau calitatea screening-ului. Optimizarea tehnicilor de analiză genetică a redus numărul de rezultate fals pozitive.
În medie, părinții copiilor care au fost testați pozitiv s-au prezentat la Centrele de referință neuromusculare la 10 zile de la nașterea copilului , cu începerea tratamentului în a 26-a zi de viață pentru cei cu două sau trei copii de SMN2. Părinții au avut preferință pentru tratamentul cu terapia genică Zolgensma (care necesită o singură administrare). Dar, potrivit autorilor, mai mult decât alegerea dintre cele trei tratamente disponibile, momentul în care se începe tratamentul (cu cât mai repede, cu atât beneficiile terapeutice sunt mai mari) este cel mai important.
Screening ul neonatal în AMS are un impact psihosocial asupra familiilor, nu numai în ceea ce privește diagnosticul, ci mai ales în ceea ce privește tratamentul, și declanșează îngrijorări cu privire la viitor, subliniind necesitatea unei îngrijiri multidisciplinare cuprinzătoare. Înțelegerea perspectivei părinților le permite consilierilor genetici să dezvolte în mod proactiv un plan de susținere și acompaniere pentru părinți în perioada dificilă de incertitudine, anxietate, frustrare și frică de necunoscut.
Surse:
https://pubmed.ncbi.nlm.nih.gov/36463459/
Editarea genetică pentru a converti gena SMN2 în genă funcțională SMN1 arată promițătoare la modele de șoareci cu AMS.
Un tratament de editare a genelor administrat într-o singură doză, care convertește gena SMN2 într-o copie funcțională a genei SMN1 a crescut nivelurile proteinei SMN care lipsește în atrofia musculară spinală (AMS), aducând o mai bună funcție motorie la modele de șoareci cu AMS, arată un studiu publicat în Science .
Combinat cu Spinraza (nusinersen), tratamentul de editare genetică a restabilit complet forța musculară, coordonarea și activitatea fizică a șoarecilor și le-a prelungit durata de viață.
„Una dintre promisiunile reale ale terapiilor de editare genetică de precizie este posibilitatea ca un tratament cu administrare unică să poată oferi o terapie pe toată durata vieții pacientului”, spune David Liu, profesor doctor la Broad Institute of Massachusetts Institute of Technology și Universitatea Harvard și autorul principal al studiului.
Mutațiile genei SMN1 sunt cauza comună a AMS, ducând la o producție redusă sau deloc de proteină SMN și la pierderea progresivă a neuronilor motori.
Genele SMN1 lipsesc sau au mutații la persoanele afectate de AMS dar au una sau mai multe copii ale genei SMN2 „de rezervă”, care codifică această proteină, iar numărul de copii ale genei SMN2 al unui pacient este, în general, asociat cu o producție mai mare de proteină SMN și un tip SMA mai ușor.
Cu toate acestea, datorită unei singure modificări a secvenței ADN a genei SMN2, celulele produc o versiune mai scurtă a proteinei SMN care se degradează rapid și nu reușește să compenseze pe deplin pierderea genei SMN1.
Cercetătorii, de la Broad Institute of MIT și Harvard, The Ohio State University Wexner Medical Center și University of Massachusetts Chan Medical School, conduși de profesorul David Liu și-au concentrat strategia pe editarea genei SMN2 pentru că au dorit să dezvolte un tratament care să funcționeze pentru toți pacienții.
Scopul echipei a fost să transforme gena SMN2 într-o copie funcțională a genei SMN1 cu o producție mai mare de proteină SMN.
„Editarea de bază este un instrument puternic pentru a corecta bolile genetice”, a spus Mandana Arbab, cercetător în laboratorul profesorului Liu și co-prim autor al studiului. „Majoritatea medicamentelor găsesc modalități de a compensa ceea ce deja nu funcționează într-o celulă, dar aici folosim editarea de bază pentru a opri AMS acolo unde își are originea, în ADN.”
Folosind o combinație de învățare automată și testare experimentală, oamenii de știință au evaluat 79 de strategii diferite de editare a nucleazei și de editare a bazelor care vizează diferite părți ale genei SMN2 și s-au concentrat pe una care a restaurat complet nivelurile proteinei SMN cu puține efecte secundare de editare și o editare redusă.. În cele din urmă, echipa a ales un editor de bază de adenină pentru a converti perechea de baze T•A din gena SMN2 într-o pereche C•G, transformând efectiv gena într-o copie funcțională a genei SMN1. Tratamentul a crescut nivelul proteinei SMN de 40 de ori, restabilindu-e la nivelurile găsite în celulele sănătoase.
Echipa de cercetare a folosit viruși folosiți în mod obișnuit în terapia genică, numiți viruși adeno-asociați (AAV) pentru a furniza editorul de bază către sistemul nervos central.
Testele au arătat că aproximativ 43% dintre neuronii motori din măduva spinării au primit moleculele necesare pentru editarea genelor, iar 87% dintre aceștia au avut conversie a genei SMN2 în genă SMN1.
Șoarecii tratați cu o singură doză de tratament de editare genetică plus Spinraza nu au arătat diferențe semnificative față de șoarecii sănătoși în ceea ce privește forța musculară, coordonarea și activitatea fizică. De asemenea, șoarecii tratați au trăit în medie de 6,5 ori mai mult decât cei fără tratament (111 față de 17 zile).
Cercetătorii au sugerat că Spinraza ar fi prelungit timpul în care neuronii motori pot supraviețui, permițând tratamentului de editare genetică mai mult timp pentru conversia genei SMN2 în genă SMN1. Deoarece această perioadă de supraviețuirea neuronilor motori este mai lungă la oameni decât la șoareci, fiind vorba de săptămâni sau luni și nu de zile ca în cazul șoarecilor, Liu a sugerat că tratamentul de editare genetică poate fi mai eficient la pacienți, în special în combinație cu terapiile pentru AMS actuale.
Laboratorul profesorului Liu dezvoltă un sistem mai simplu de “livrare “ a tratamentului cu un singur adenovirus asociat (AAV), care ar putea reduce doza și ar putea simplifica abordarea tratamentului . Prof. Liu speră că sistemul de livrare îmbunătățit ar putea oferi o cale către studiile clinice.
Surse:
SMA tip I: dilema părinților raportată la decizia de administrare a tratamenteor inovative
Decizia părinților copiilor nou diagnosticați cu SMA de a administra și de a alege un medicament inovativ la care nu cunoaștem încă nici toate efectele, nici eficacitatea reală, în special pentru un copil cu SMA tip I este una dificilă, personală și complexă,decizie care trebuie să beneficiaze de suport psihologic adecvat.
Un studiu efectuat în Franța, a căutat să înțeleagă mai bine experiența părinților atunci când decid să opteze pentru un tratament inovativ (Spinraza®, Zolgensma®, Evrysdi®) sau îngrijiri paleative pentru copilului lor afectat de amiotrofie musculară spinală (SMA) tip I, în special dacă copilul nu prezintă simptome sau, din contră, simptomele sunt deja foarte severe și să evalueze consecințele psihologice pe termen mediu în viața lor ca părinți.
Pentru a face acest lucru, optsprezece părinți (12 mame și 6 tați) a treisprezece copii cu SMA tip I tratați cu un tratament inovativ au avut un interviu cu un psiholog și au răspuns la un auto-chestionar la puțin peste doi ani.în medie, după inițierea tratamentului.
Decizia trebuie să fie una informată dar rapidă
Majoritatea părinților au relatat că au simțit că au fost implicați de echipa medicală atunci când au decis alegerea tratamentului pentru copil și că punctul lor de vedere a fost luat în considerare.
Mai mulți părinți au căutat informații si de la asociațiile de pacienți care le-au permis să-și facă o idée mai clară despre tratamente sau despre îngrijirile paliative prin experiența altor părinți. De asemenea, le-a permis să aibă o a doua opinie de la un alt centru de expertiză atunci când nu au fost convinși sau când le-au fost propuse doar îngrijiri paleative.
În medie, majoritatea părinților au fost mulțumiți de decizia luată și de progresul copilului lor sub tratament. Cu toate acestea, cinci părinți au fost mai puțin mulțumiți din cauza unor probleme medicale la inițierea tratamentului și/sau a progresulor mai mici decât s-au așteptat și/sau a apariției semnelor bolii.
Aflarea diagnosticului i-a pus pe părinți în fața unui viitor marcat fie de un deces precoce, fie de un handicap sever și singura posibilitate de a acționa era să ia o decizie.
Decizia tratamentului inovativ rămâne permanentă pe parcursul evoluției copilului; în special, este pusă sub semnul întrebării de fiecare dată când starea de sănătate a copilului se deteriorează și se confirmă riscul unui handicap grav. Patru dintre părinții participanți își fac griji că copilul lor îi va învinovăți într-o zi pentru decizia lor.
La momentul deciziei, niciunul dintre părinți nu a simțit că a înțeles pe deplin nici toate implicațiile bolii și nici efectele benefice sau adverse ale tratamentelor.
Mai mulți părinți declară că și-au construit o reprezentare idealizată pe baza expunerii medicului, a informațiilor găsite pe internet și/sau obținute de la asociațiile de pacienți, precum și a dorinței de a-și salva copilul.
Deși nu există date despre toleranța și eficacitatea sa pe termen lung, terapia genică a părut familiilor a fi mai eficientă și mai puțin traumatizantă decât alte tratamente.
În general, părinții participanți nu au fost depresivi și au avut un nivel scăzut de anxietate.Un grup de 11 părinți „neezitanți” în luarea uneidecizii era mai deprimat și mai anxios decât cel al restului de șapte părinți „ezitanți”, care nu erau sau foarte puțini deprimați sau anxioși, sugerând că ezitarea în luarea deciziei nu predispune neapărat la depresie sau anxietate.
Deși rezultatele acestui studiu de mică anvergură nu pot fi generalizate, ele arată că în fața provocărilor pe care această boală le impune părinților unui copil nou diagnosticat cu SMA, fiecare experiență este un proces intim influențat de istoria personală a fiecăruia. Acest proces decizional este determinat de dorința de a oferi copilului toate șansele la o viață normală , de încrederea în medicul prescriptor și de nevoia de a salva viața copilului .
Urgența deciziei trebuie să încurajeze echipa medicală să ofere sistematic părinților o întâlnire cu un psiholog, dacă este posibil cu o bună cunoaștere a SMA tip I, atât în timpul procesului luării deciziei,cât și după luarea acesteia.
Sursă :
O nouă pistă terapeutică pentru AMS
https://medicalxpress.com/news/2023-03-insights-spinal-muscular-atrophy.html
Protocoalele terapeutice aferente tratamentelor AMS și lista centrelor acreditate în derularea programului național de boli rare- componenta P6.24 publicate în monitorul oficial și pe site-ul CNAS.
ZOLGESMA
Noi date privind urmărirea pe termen lung a pacienților prezentate la conferința MDA
EVRYSDY
Date noi la patru ani pentru Evrysdi de la Roche consolidează eficacitatea pe termen lung și profilul de siguranță la unele dintre persoanele cele mai grav afectate de atrofia musculară spinală tip 2 și 3
https://ml-eu.globenewswire.com/Resource/Download/7d888e5c-14ad-4e85-bb9d-fdc00d5e5724
Zolgesma (ONASEMNOGEN ABEPARVOVEC) accesibil pentru pacienții români eligibili conform protocolului terapeutic.
Ordinul ministrului sănătății și al președintelui Casei
Naționale de Asigurări de Sănătate nr. 689/157/2023
privind modificarea și completarea anexelor nr. 1 și 2
la Ordinul ministrului sănătății și al președintelui Casei
Naționale de Asigurări de Sănătate nr. 564/499/2021
pentru aprobarea protocoalelor terapeutice privind
prescrierea medicamentelor aferente denumirilor
comune internaționale prevăzute în Lista cuprinzând
denumirile comune internaționale corespunzătoare
medicamentelor de care beneficiază asigurații, cu sau
fără contribuție personală, pe bază de prescripție
medicală, în sistemul de asigurări sociale de sănătate,
precum și denumirile comune internaționale
corespunzătoare medicamentelor care se acordă în
cadrul programelor naționale de sănătate, aprobată
prin Hotărârea Guvernului nr. 720/2008, și a normelor
metodologice privind implementarea acestora a fost publicat în Monitorul Oficial .
Mulțumim tuturor instituțiilor, Agenția Națională a Medicamentului și a Dispozitivelor Medicale din România, Ministerul Sănătății – România, CNAS – Casa Națională de Asigurări de Sănătate și tuturor factorilor implicați care au făcut posibil ca toate tratamentele pentru aprobate pentru atrofia musculară spinală să fie accesibile pacienților români afectați de această maladie.
Contăm mai departe pe sprijinul instituțiilor, comisiilor de specialitate, societăților profesionale medicale, asociațiilor de pacienți, medicilor, pacienților și părinților pentru implementarea standardelor de îngrijire specifice pentru această boală și a unui program național de screening neonatal pentru AMS.
SMA tip I și nusinersen: efect redus sau deloc asupra scoliozei ?!
Șapte sugari afectați de atrofie musculară spinală tip I tratați cu nusinersen înainte de vârsta de 6 luni, au dezvoltat toți scolioză înainte de vârsta de un an.
Aceasta este experiența unei echipe canadiene, care i-a urmărit pe acești șapte copii timp de 10 până la 57 de luni. În medie, în timpul perioadei de studiu au fost administrate nouă doze (între 5 și 15) de nusinersen per copil.
Analiza performanțelor motorii ale copiilor arată o îmbunătățire mai ales la nivelul membrelor, dar nu și a musculaturii implicate în menținerea capului și a trunchiului. Toți copiii au dezvoltat scolioză înainte de vârsta de un an din cauza slăbiciunii musculare la nivelul spatelui, chiar dacă li s-a administrat nusinersen.
După cum au sugerat și alte studii, în atrofia musculară spinală de tip I , nusinersen pare să fie mai eficient asupra mușchilor membrelor decât asupra mușchilor trunchiului și gâtului, deși motivul nu este încă clar.
Sursă:
Pentru prima dată, un studiu de istorie naturală la adulți cu AMS arată diferențe în funcție de sex: boala ar fi mai gravă la bărbații adulți decât la femei.
Până în prezent există puțini factori care să estimeze evoluția atrofiei musculare spinale . Cel mai frecvent utilizat este numărul de copii ale genei SMN2, dar poate avea anumite limitări.
Pentru a avea mai mulți factori predictivi, doi clinicieni italieni au analizat datele clinice și moleculare a 165 de adulți cu SMA (64 femei pentru 101 bărbați), 13% SMA tip II, 85% tip III și 2% tip IV.
Rezultatele nu au arătat diferențe semnificative între bărbați și femei în distribuția după tipul de SMA și nici în numărul mediu de copii ale genei SMN2.
La bărbați
Pe de altă parte, ei arată pentru prima dată un efect în funcție de sex asupra funcției motorii a bolii, cu o severitate mai mare a bolii la bărbați, și mai ales la adulții tineri cu SMA de tip III, decât la femei.
La femei
Conform studiului, boala a apărut mai devreme la femei (la vârsta de 3 ani) decât la bărbați (4 ani). Pe de altă parte, femeile cu SMA tip III cu 3 sau 4 copii de SMN2 mențin mersul mai mult decât bărbații.
Cu toate acestea, vor fi necesare cercetări suplimentare pentru a confirma sau nu aceste noi observații.
Sursă:
Un studiu din viața reală arată stabilizarea sau chiar îmbunătățirea forței motorii la persoanele non-ambulante afectate de SMA de tip II sau III cărora li se administrează nusinersen.
Datele din viața reală au fost colectate ca parte a registrului SMArtCARE, de la 256 de persoane cu atrofie musculară spinală (AMS) de tip II sau III, non-ambulante și tratate cu nusinersen (Spinraza®).
După o perioadă de un an (pentru aproape 90% dintre ei) până la trei ani (pentru jumătate dintre ei) de tratament, funcția motrică a rămas stabilă în 70% din cazuri și s-a îmbunătățit la un sfert dintre participanți. În aproape toate cazurile, forța brațului nu s-a deteriorat. S-a îmbunătățit în aproape o treime dintre ei, inclusiv la cei cu vârste mai mari.
Sursă:
Va invit sa urmăriți un interviu despre diagnostic, ce înseamnă amiotrofia spinala si importanta tratamentului timpuriu.
Sa nu luati in considerare emotiile foarte mari :)
O seara buna va doresc!
Noile date din studiul de fază 2 TOPAZ, indică tendințe pozitive în măsurarea calității vieții de-a lungul a 24 de luni de tratament cu apitegromab pentru pacienții nonambulatori afectați de SMA tip 2 și 3.
https://investors.scholarrock.com/…/new-phase-2-topaz…
Lansare site https://www.semnealeams.ro/
Agenția Europeană a Medicamentului a actualizat prospectul pentru Everysdi (Risdiplam).
Dacă până acum medicamentul trebuia păstrat în frigider tot timpul, acum poate fi păstrat la temperatura camerei ( sub 40°C) până la 120 de ore (5 zile) în total.
Această actualizare e binevenită mai ales pentru călătorii.
O doza mai mare de Spinraza poate fi mai eficientă, după un nou studiul de modelare.
Cu bucurie vă anunțăm că începând de azi Everysdi (Risdiplam ) este oficial în lista medicamentelor compensate .
Termenul prevăzut de legislație pentru elaborarea protocolului terapeutic de către comisia de neurologie pediatrică este de maxim 30 de zile și vom face tot ceea ce depinde de noi ca acest termen să fie respectat. Compania farmaceutică care deține autorizația de punere pe piață a început deja negocierile cu CNAS privitoare la bugetarea acestei terapii și sperăm ca în cel mai scurt timp pacienții eligibili să poată beneficia de această moleculă.
În ceea ce privește Zolgesma , în ciuda unei opoziții puternice, vom face toate demersurile legale posibile astfel încât la următoarea actualizare a HG 720/2008 terapia genică pentru AMS să fie inclusă în listă.
https://drive.google.com/file/d/1MizGr014o6NGA-j3oFSmoPNywf-72PiR/view
În amiotrofia musculară spinală nelegată de deleția genei SMN1 se pare că gena CAPN1(gena care codifică calpaina 1) ar putea fi una dintre cauze.
Dacă 96% din cazurile de atrofie musculară spinală (SMA) se datorează unor anomalii ale genei SMN1, există alte forme de SMA, adesea diferite clinic. De asemenea, se întâmplă ca formele care evocă atrofia musculară spinală să aibă alte caracteristici genetice.
Acest lucru a fost raportat de o echipă australiană care nu a găsit o anomalie a genei SMN1 la un frate și o soră care, totuși, au prezentat slăbiciune a umerilor și brațelor, pelvisului și coapselor apărute la vârsta adolescenței, manifestări similare cu cele ale SMA tip IV.
Analiza întregului lor genom a relevat o anomalie în gena CAPN1, care codifică calpaina1.
Aceasta este o enzimă care, sub influența calciului, distruge proteinele (proteaza). Se găseste in toate tesuturile corpului, spre deosebire de calpaina 3, specifica muschiului si a carei deficienta duce la distrofia musculara a membrelor.
Studiul anumitor celule prelevate de la fratele si sora în cauză, fibroblastele, a confirmat scăderea cantitații si activitații calpainei1.
Sursă:
Noul studiu STRENGTH (Zolgesma /Novartis) așteptat să înceapă în a doua parte a anului pentru cei tratați în prezent cu Spinraza sau Everysdi (2-12 ani) și care vor să treacă pe Zolgesma ( administrare intratecală ). Studiu open label deci fără placebo.
Aprobarea Zolgesma prin administrare intratecală se speră să aibă loc în 2025.
https://drive.google.com/file/d/14TPFC5sNAeS50M-KhzMEedR1WXq1zOIX/view
Evrysdi (risdiplam) de la Roche a primit evaluarea prioritară a FDA pentru tratamentul bebelușilor presimptomatici sub două luni cu atrofie musculară spinală (SMA)
O nouă pistă terapeutică în SMA
Oligonucleotidele antisens care vizează regiunile metilate ale genei SMN2 ar permite producerea unei proteine SMN funcționale la șoareci și in vitro.
Scopul tratamentelor actuale pentru atrofia musculară spinală (SMA) este de a crește producția de proteină SMN. Abordări precum corectarea splicing-ului genei SMN2 sau stimularea transcripției acesteia merg în această direcție.
Expresia genei SMN2 este reglată în special de modificări epigenetice, cum ar fi metilarea. Când aceasta afectează regiunea de început a transcripției genei (regiunea promotoare), producția de proteină SMN este redusă sau reprimată. Cu cât nivelul de metilare este mai mare, cu atât producția de SMN este reprimată și forma de SMA mai severă. Acționarea asupra acestor regiuni metilate pentru a crește producția de proteină SMN ar reprezenta o nouă abordare terapeutică.
Așa-numiții factori „epigenetici” reglează informația genetică acționând asupra organizării moleculei de ADN (mai mult sau mai puțin condensată, metilată sau nu etc.) și nu asupra conținutului acesteia (secvența sa de nucleotide este conservată). O modificare epigenetică a expresiei genei poate apărea spontan, ca răspuns la mediu, și poate fi reversibilă. Se poate transmite și în timpul diviziunii celulare.
Un studiu din China a analizat, în celule derivate de la persoane cu SMA și la șoareci , efectele administrării a două oligonucleotide antisens care vizează regiunile cheie de metilare ale SMN2. O creștere semnificativă a producției de proteină SMN a fost observată după adăugarea oligonucleotidelor. Autorii au obținut rezultate în celulele SMA tratate similare cu nusinersen (Spinraza®), o altă oligonucleotidă antisens care acționează asupra splicing-ului SMN2.
Administrarea intratecală a uneia dintre cele două oligonucleotide antisens (ASO-P1) la șoareci ,a crescut nivelul proteinei SMN în diferite organe (mușchi, creier, măduva spinării, ficat) și durata de viață a acestora.
În acest studiu, tratamentul combinat nusinersen și noua oligonucleotidă ASO-P1 a crescut nivelul SMN funcțional în celule nusinersen singur sau mai mult decât ASO-P1 singur .
Administrarea simultană a celor două oligonucleotide ar putea crește nivelul proteinei SMN.